Product Overview
Blue-light cystoscopy (BLC), also referred to as fluorescent cystoscopy or photodynamic diagnosis (PDD), is a procedure in which a photosensitizer medication is instilled in the bladder prior to cystoscopy. This photosensitizer is part of the heme biosynthesis pathway (that makes red blood cells) and causes an accumulation of photoactive porphyrins in neoplastic cells. These porphyrins preferentially accumulate in neoplastic cells due to the increased metabolic activity in these cells. When excited with blue-light in the 360-450 nm wavelength, the porphyrins emit a red light that can easily be seen during cystoscopy (Figure 1). There are two main photosensitizers that have been used in studies looking at fluorescent cystoscopy: 5-aminolevulinic acid (5-ALA) and hexaminolevulinate (HAL). HAL is the only photosensitizer that has been approved for use in the USA and Europe. In the USA it is marketed under the brand name Cysview, and in Europe under the brand name Hexvix. |
| Figure 1. 63 year old female with prior TURBT+BCG now with recurrence. (a) WLC of a prior resection site near a ureteral orifice. (b) Same site using BLC. Tumor was found to be high grade T1. (c) WLC showing an area of CIS that was missed upon repeat resection. (d) Same site using BLC. From: Daneshmand, et al.[2] |
Increased Tumor Detection
Most studies are in agreement that more tumors are found when using BLC compared to WLC alone. Perhaps the best evidence for this is a meta-analysis published in 2013 which looked at the raw data from 6 prospective studies including a total of over 800 patients[3]. This meta-analysis found that a significant proportion of tumors were missed by WLC alone. In fact, an additional 14.7% of Ta tumors were detected with BLC, 10.8% of T1 tumors, and 40.8% of CIS (carcinoma in situ or flat tumors) (Figure 2). Another meta-analysis by Shen et al4. failed to show a statistically significant difference in tumor detection rates between BLC and WLC, however this study was limited by the inclusion of a large number of studies which used the photosensitizer 5-ALA, which is not FDA approved and has been shown to have less fluorescent properties than HAL.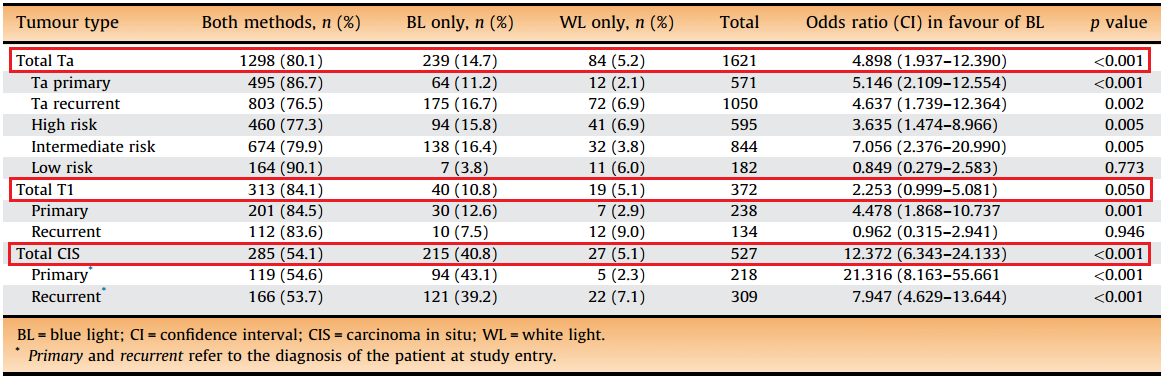 |
| Figure 2. Increased detection of tumors using BLC alone when compared to WLC alone. From: Burger, et al.[3] |
Recurrence Free Survival
While the data on increased detection are consistent, data on recurrence-free survival (RFS) are less clear-cut. Cysview gained FDA approval following a study by Stenzl et al5. published in 2010. This study was a prospective, randomized, multi-center study that looked at 551 patients with suspected Ta or T1 disease and were randomized to either WLC or WLC+BLC. During the 9-month surveillance period, 47% of patients in the BLC group and 56% of patients in the WLC group had tumor recurrences for a relative reduction of 16%. Interestingly, the following year Stenzl published another paper which randomized patients to either 5-ALA or placebo which failed to show a difference in recurrence-free survival 12 months after tumor resection6. However, as previously mentioned, this study used 5-ALA which is not the FDA approved photosensitizer.The study with the longest follow-up data is actually an extension to the original Stenzl study used for FDA approval. This study was published in 2012 by Grossman et al7. and showed that with a median follow-up for 53.0 months (WLC group) and 55.1 months (BLC group), 38% of the patients in the BLC group remained tumor free vs. 31.8% in the WLC group. The median time to recurrence was 16.4 months in the BLC group and 9.4 months in the WLC group. This study also looked at progression-free survival and cystectomy rates but was unable to show a statistical difference between the two groups, possibly due to the original study not being powered to look at differences in these outcomes.
Figure 3 summarizes additional studies that have looked at recurrence-free survival for BLC vs. WLC.
 |
| Figure 3. Summary of studies that have looked at recurrence-free survival for BLC vs. WLC. |
Cost
Bladder cancer is one of the mostly costly cancers to treat on a per capita basis. Lifetime per capita costs have been estimated between $96,000 and $187,000 (2001 US dollars)8. Multiple analyses have looked at whether using BLC could reduce cost for bladder cancer treatments using the assumption that if a patient has a longer recurrence-free survival, they may require fewer or perhaps less frequent TURBTs. Garfield et al9. used a probabilistic decision-tree model and estimated that over 5 years, approximately $4,600 could be saved per patient by using BLC during diagnostic cystoscopy (excluding the cost of the equipment).Indications
The current AUA guidelines for bladder cancer were written in 2007, 3 years before Cysview was approved for use in the USA and therefore do not have an official recommendation for the use of fluorescent cystoscopy in the management of NMIBC. The NCCN guidelines acknowledge that BLC has been shown to decrease recurrence in NMIBC but has not been shown to reduce progression. They suggest that “BLC may have the greatest advantage in detecting difficult-to-visualize tumors (eg, CIS tumors)” and “the limitations of BLC require judicious application of this additional diagnostic tool”10.In 2013, an expert focus group convened in San Diego to create a consensus statement for appropriate use of BLC in the USA2. Based on the evidence reviewed, they recommend that BLC should be considered:
- At initial TURBT on suspicion of NMIBC
- In patients with positive urine cytology but negative WLC findings
- In patients with intermediate-risk NMIBC
- For assessment of disease recurrence
These recommendations are similar to consensus statements and guidelines from Europe.
Summary
Blue-light cystoscopy has been shown to increase detection of NMIBC during TURBT. Whether this increased detection leads to a difference in recurrence or progression is less clear. Most studies to date, especially those with longer follow-up times, have shown a decrease in recurrence in patients who undergo TURBT with BLC. No study to date has shown a difference in progression. It is likely that to show a statistically significant difference in progression, larger studies with longer follow-up will need to be conducted.
REFERENCES
1. Cancer of the Urinary Bladder - SEER Stat Fact Sheets. http://seer.cancer.gov/statfacts/html/urinb.html. Accessed September 2, 2015.
2. Daneshmand S, Schuckman AK, Bochner BH, et al. Hexaminolevulinate blue-light cystoscopy in non-muscle-invasive bladder cancer: review of the clinical evidence and consensus statement on appropriate use in the USA. Nat Rev Urol. 2014;11(10):589-596. doi:10.1038/nrurol.2014.245.
3. Burger M, Grossman HB, Droller M, et al. Photodynamic diagnosis of non-muscle-invasive bladder cancer with hexaminolevulinate cystoscopy: a meta-analysis of detection and recurrence based on raw data. Eur Urol. 2013;64(5):846-854. doi:10.1016/j.eururo.2013.03.059.
4. Shen P, Yang J, Wei W, et al. Effects of fluorescent light-guided transurethral resection on non-muscle-invasive bladder cancer: a systematic review and meta-analysis. BJU Int. 2012;110(6 Pt B):E209-E215. doi:10.1111/j.1464-410X.2011.10892.x.
5. Stenzl A, Burger M, Fradet Y, et al. Hexaminolevulinate guided fluorescence cystoscopy reduces recurrence in patients with nonmuscle invasive bladder cancer. J Urol. 2010;184(5):1907-1913. doi:10.1016/j.juro.2010.06.148.
6. Stenzl A, Penkoff H, Dajc-Sommerer E, et al. Detection and clinical outcome of urinary bladder cancer with 5-aminolevulinic acid-induced fluorescence cystoscopy : A multicenter randomized, double-blind, placebo-controlled trial. Cancer. 2011;117(5):938-947. doi:10.1002/cncr.25523.
7. Grossman HB, Stenzl A, Fradet Y, et al. Long-term decrease in bladder cancer recurrence with hexaminolevulinate enabled fluorescence cystoscopy. J Urol. 2012;188(1):58-62. doi:10.1016/j.juro.2012.03.007.
8. Botteman MF, Pashos CL, Redaelli A, Laskin B, Hauser R. The health economics of bladder cancer: a comprehensive review of the published literature. Pharmacoeconomics. 2003;21(18):1315-1330. http://www.ncbi.nlm.nih.gov/pubmed/14750899. Accessed September 2, 2015.
9. Garfield SS, Gavaghan MB, Armstrong SO, Jones JS. The cost-effectiveness of blue light cystoscopy in bladder cancer detection: United States projections based on clinical data showing 4.5 years of follow up after a single hexaminolevulinate hydrochloride instillation. Can J Urol. 2013;20(2):6682-6689. http://www.ncbi.nlm.nih.gov/pubmed/23587507. Accessed September 2, 2015.
10. NCCN Clinical Practice Guidelines in Oncology: Bladder Cancer. 2015;http://www. http://www.nccn.org/professionals/physician_gls/PDF/bladder.pdf. Accessed September 2, 2015.











